
Before MoCRA, the U.S. cosmetics industry operated under a regulatory framework that had not been meaningfully updated since 1938. Product facility registration was voluntary. Adverse event reporting was not required. FDA had no mandatory recall authority. Quality standards were guidelines, not enforceable requirements. The result was an industry where failure investigation and CAPA systems – where they existed at all – were designed for internal quality management rather than regulatory compliance. That is no longer the case.
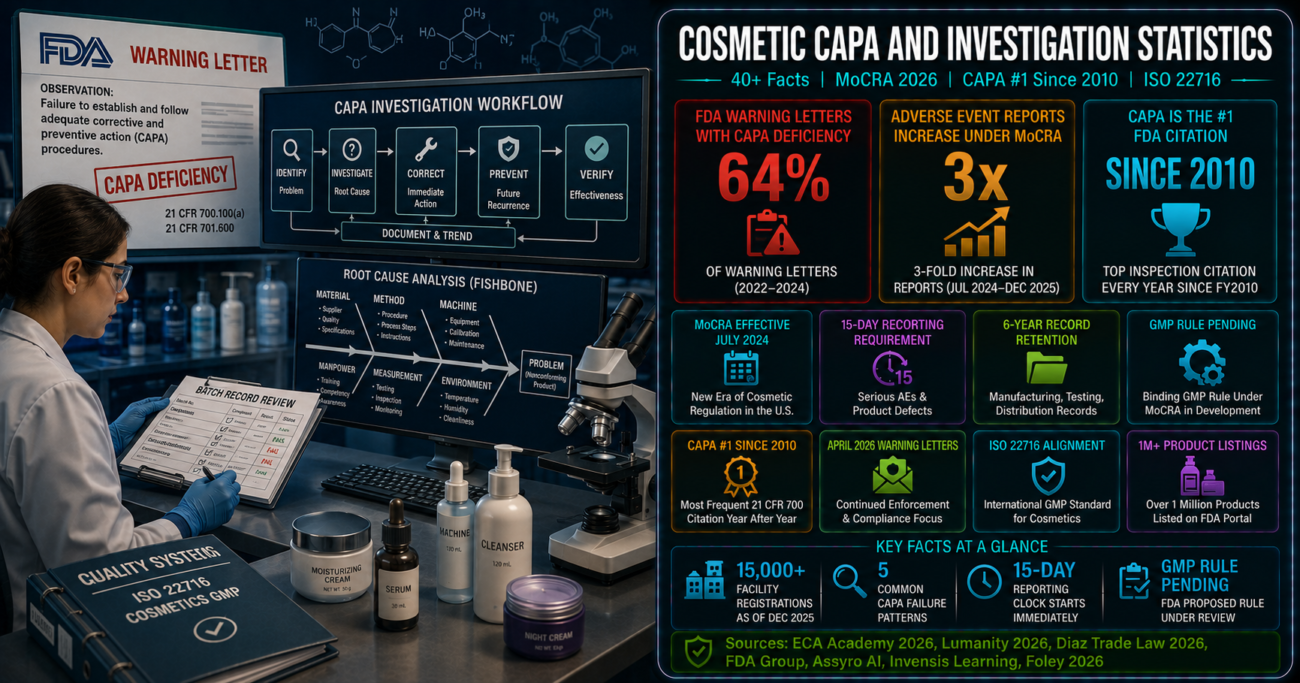
MoCRA’s serious adverse event reporting requirement, its six-year record retention mandate, its mandatory facility registration and product listing (enforced from July 2024), and the forthcoming binding GMP regulation mean that cosmetic manufacturers must now maintain investigation and CAPA programs that can withstand FDA scrutiny. The playbook for what that scrutiny looks like is well-documented in the pharmaceutical CAPA enforcement record: CAPA has been the top FDA inspection citation every year since 2010, appearing in 64% of warning letters between 2022 and 2024. The failures FDA cites are consistent, predictable, and preventable. Below we compile 40+ statistics on MoCRA’s enforcement posture, CAPA failure patterns from FDA’s own warning letters, root cause analysis methodology, and what effective investigation programs look like in 2026.
Editor's Choice: Key CAPA and MoCRA Statistics for Cosmetic Manufacturers
1. MoCRA in 2026: What Has Changed and What Is Still Coming
- MoCRA is the most significant change to U.S. cosmetic regulation since the FD&C Act of 1938 – a period of over 80 years during which cosmetics were regulated under a framework fundamentally unchanged. The law broadens FDA’s authority, expands industry obligations, and is reshaping how cosmetics are brought to market, monitored, and recalled. (National Law Review, May 2026; Foley and Lardner, May 2026)
- FDA has already achieved significant visibility into the cosmetics market through MoCRA’s registration and listing requirements: as of mid-2026, the FDA database shows 15,000 unique active facility registrations and over one million unique active cosmetic product listings. This is the first comprehensive FDA map of the U.S. cosmetics manufacturing landscape – and it is the database that will drive inspection targeting under Elsa and other risk-based systems. (FDA / Diaz Trade Law, May 2026)
- Adverse event reports from the cosmetics industry have increased by more than threefold since MoCRA’s mandatory serious adverse event reporting requirement took effect. This surge is a direct consequence of mandatory reporting replacing a voluntary system – the underlying event rate has not necessarily increased, but the reporting rate has, creating a much richer signal for FDA enforcement targeting. (FDA / Diaz Trade Law, May 2026)
- FDA is expected to continue increasing its cosmetic enforcement activity throughout 2026 and beyond. The National Law Review (May 2026) and Foley and Lardner (May 2026) both note that FDA is sharpening its focus on high-risk cosmetic ingredients and using the new public product listing database to drive transparency and, indirectly, enforcement. The combination of mandatory listing data and Elsa AI targeting means high-risk facilities can be identified before an inspector arrives. (National Law Review, May 2026)
2. How CAPA Fails: The Patterns FDA Cites in Warning Letters
- CAPA has been the number one FDA inspection citation every fiscal year since 2010 – a 15-year consecutive run. According to FDA Group’s warning letter analysis cited by Invensis Learning (May 2026), CAPA deficiencies have appeared consistently in the top 10 inspection observations for this entire period, reflecting a structural gap rather than a cyclical enforcement priority. (FDA Group; Invensis Learning, May 2026)
- Inadequate CAPA systems appeared in 64% of FDA enforcement actions against drug manufacturers in 2024 alone, second only to validation failures. While these figures come from the drug manufacturing enforcement record, cosmetic GMP requirements under MoCRA are expected to generate similar enforcement patterns once the binding GMP rule takes effect. (Assyro AI / FDA analysis, March 2026)
- ECA Academy’s May 2026 analysis of FDA’s April 2026 warning letters is precise about what FDA expects: “investigations must be thorough, well-documented, scientifically sound, and timely, and they must translate into CAPA that is appropriately scoped and effective.” The failure chain FDA cites is when any link in this sequence breaks – shallow investigation, inadequate scope, training-only CAPA, or no effectiveness verification. (ECA Academy, May 2026)
- ECA Academy’s September 2025 analysis of four warning letters published in that period found a specific pattern: microbial deviations not adequately investigated, with products released for distribution despite evidence of objectionable organisms. In one case, a second positive finding for the same organism triggered only a retest and partial rejection – not a root cause investigation of why the contamination recurred. This is the investigation failure that precedes the CAPA failure. (ECA Academy, September 2025)
- The FDA Group’s audit intelligence (January 2025) identifies the fundamental problem with most CAPA systems: teams treat CAPA as a compliance requirement rather than an actual problem-solving tool. This mindset creates predictable cascading failures: every deviation becomes a potential CAPA (overwhelming the system); documentation becomes more important than resolution; teams focus on closing CAPAs rather than preventing recurrence; root cause analysis becomes a checkbox exercise; effectiveness checks measure completion rather than improvement. (The FDA Group, January 2025)
3. The Five Most Common CAPA Deficiencies in FDA Observations
- These five deficiency categories appear across FDA’s enforcement record for drug, device, and OTC product manufacturers – and will apply equally to cosmetic manufacturers once binding GMP regulations are finalized under MoCRA. The underlying failure dynamics are structural, not industry-specific: they reflect how quality management systems behave when compliance pressure dominates over problem-solving culture. (FDA Group; Invensis Learning, 2026)
- For cosmetic manufacturers, the most operationally critical of the five deficiencies is trending failure. Under MoCRA’s mandatory adverse event reporting, FDA will have access to reported complaint and adverse event data that cosmetic manufacturers may not be systematically analyzing for internal trends. An FDA investigator who has identified a clustering pattern across adverse event reports before the inspection – and finds the manufacturer’s CAPA program has not detected that same pattern internally – has a clear basis for citing both the investigation failure and the CAPA deficiency. (MoCRA adverse event data implications; FDA targeting analysis)
4. Root Cause Analysis Tools: What Works and When to Use Each
- The fundamental principle underlying all effective root cause analysis tools is the same: a CAPA is only as good as the root cause analysis that feeds it. The most common reason CAPA fails – per all major quality system analyses – is that the investigation did not identify the true root cause before the corrective action was designed. An action plan built on an incorrect or incomplete root cause will not prevent recurrence regardless of how well-documented it is. (JJC Group, December 2025; ECA Academy, 2026)
- The 5 Whys is the most widely deployed root cause tool but also the most commonly misapplied. Teams that stop at two or three “why” iterations, or that follow the most obvious causal path while ignoring parallel causal chains, produce root cause conclusions that identify symptoms rather than system failures. For cosmetic manufacturing failures involving microbial contamination, formulation deviations, or equipment performance, 5 Whys must be applied in conjunction with scientific data – not as a substitute for laboratory investigation. (SmartCAPA; FDA Group, 2025)
- FMEA has specific relevance for cosmetic manufacturers preparing for MoCRA’s GMP rule. Applying FMEA to existing manufacturing processes identifies failure modes that should be formally assessed for their RPN before an inspection – or before a complaint signals that the failure mode has realized. FMEA used reactively after an incident can evaluate whether the failure mode was known and what controls were assumed to prevent it. (SmartCAPA; Center for Professional Innovation and Education, 2025)
- The FDA Group’s January 2025 CAPA tips specifically recommend including both quality representatives and experienced operators in the initial investigation assessment. Operators often identify subtle changes in equipment behavior that preceded the deviation, while quality personnel verify documentation and regulatory compliance. Excluding production knowledge from the investigation consistently produces incomplete root cause conclusions. (The FDA Group, January 2025)
5. MoCRA Investigation Triggers: What Requires a Formal Investigation Under the New Framework
- MoCRA defines “serious adverse event” as any event associated with a cosmetic product that results in (1) death, (2) serious injury (hospitalization, persistent or significant disability, congenital anomaly, or risk of death), or (3) any adverse event that requires inpatient hospitalization, medically important event, or other events of a serious nature. The 15-business-day clock begins when the responsible person (manufacturer, packer, or distributor) first receives the report – not when they determine the event is “probably” product-related. (MoCRA Section 605; FDA guidance)
- FDA’s mandatory recall authority under MoCRA is new. Prior to MoCRA, recalls were voluntary; FDA could only request that a manufacturer recall and had to pursue court action to compel it. Mandatory recall authority fundamentally changes the investigation dynamic: a manufacturer that cannot produce adequate investigation documentation supporting a distribution suspension or recall decision faces both a product safety risk and a new enforcement mechanism that did not exist before 2022. (Lumanity, May 2026; MoCRA)
- MoCRA’s records access authority allows FDA to request records relevant to cosmetic product safety. Six-year record retention is mandatory under Section 605. Investigation records, CAPA documentation, adverse event reports, and complaint files that are missing, incomplete, or produced in non-retrievable formats create an enforcement exposure independent of the underlying quality event they were meant to document. (MoCRA Section 605; Respect Manufacturing, 2026)
- For OTC cosmetic products – sunscreens, acne treatments, dandruff shampoos – the investigation framework combines MoCRA’s cosmetic requirements with OTC drug manufacturing standards. Active Cosmetics Manufacturing Inc. received one of FDA’s April 2026 warning letters specifically because its investigation of a contamination event did not extend to other lots made under the same conditions – a finding that applies equally to cosmetic-only manufacturers preparing for MoCRA GMP compliance. (ECA Academy, May 2026)
6. Preparing for the MoCRA GMP Rule: What ISO 22716 Requires Now
- MoCRA mandates that FDA issue binding GMP regulations for cosmetics. The rule was expected by late 2025 but has not been finalized as of mid-2026. ISO 22716 is the international cosmetics GMP standard and the most likely model for what the final rule will require. Companies implementing ISO 22716 now are building the compliance infrastructure that will be mandatory once the rule is finalized – rather than facing a remediation cycle after enforcement begins. (National Law Review, May 2026; Respect Manufacturing, 2026)
- ISO 22716 Section 4.8 requires that cosmetic manufacturers establish procedures for identifying, investigating, and correcting nonconformities, and for preventing their recurrence through CAPA. The documentation requirement is explicit: investigation findings and corrective actions must be documented. This requirement is substantively aligned with what FDA expects in the pharmaceutical and OTC drug context, and will generate enforcement expectations for cosmetics once binding. (ISO 22716; industry analysis)
- FDA’s guidance for MoCRA compliance emphasizes that safety substantiation data, batch records, and traceability logs that prove product safety must be maintained. These are the same records that support investigation and CAPA programs: batch records establish the conditions under which nonconformances occurred; traceability logs identify affected lots; safety substantiation provides the scientific basis for assessing whether a deviation constitutes a safety risk. (MoCRA guidance; Respect Manufacturing, 2026)
- FDA moved the Office of Cosmetics and Colors from CFSAN to the Office of the Chief Scientist in 2024 “to better align with its core mission” – a structural signal of elevated institutional focus on cosmetics. Combined with the Cosmetics Direct database, the Elsa AI targeting system, and MoCRA’s new enforcement authorities, the cosmetics inspection environment of 2026 is materially more demanding than 2021. (Lumanity, May 2026)
7. What an Effective Cosmetic Manufacturer CAPA Program Looks Like in 2026
- Effective CAPA programs for cosmetic manufacturers in 2026 start with risk-based triage: a documented, objective system for determining whether an event requires a full CAPA or a correction. Applying CAPA to every minor deviation overwhelms the system and produces superficial investigations across the board. Applying CAPA only to the most serious events without a documented triage rationale creates the same FDA exposure as having no process at all. (The FDA Group, January 2025; quality system best practices)
- ECA Academy’s May 2026 analysis articulates FDA’s requested remediation pattern from the April 2026 warning letters: independent retrospective reviews, stronger investigation competencies, better root cause evaluation, CAPA effectiveness verification, and stronger Quality Unit oversight. Note the order: the investigation precedes the CAPA, and the Quality Unit oversight frames both. Effective programs ensure each element is in place before the investigation opens, not assembled reactively after FDA issues observations. (ECA Academy, May 2026)
- For MoCRA-specific adverse event investigation, the most critical operational requirement is speed combined with completeness. The 15-business-day reporting clock means cosmetic manufacturers cannot afford the multi-week investigation timelines common in pharmaceutical settings for routine deviations. Investigation SOPs must be pre-designed with templates, responsibility assignments, and documentation frameworks that allow rapid but rigorous initial assessment within days of event receipt. (MoCRA 15-day requirement; investigation program design)
- Trending is the quality system capability most likely to differentiate cosmetic manufacturers that are ready for MoCRA enforcement from those that are not. FDA now has access to the industry’s adverse event reporting data through MoCRA’s reporting requirement. An investigator who identifies a signal in that data that the manufacturer’s own trending program has not detected is observing a failure of the quality system, not a reporting gap. Monthly review of adverse events and complaint data by category, ingredient, and product line is the minimum trending frequency for cosmetic manufacturers with significant product volumes. (MoCRA adverse event data; quality trending best practices)
Key Takeaways for Cosmetic Manufacturers and Quality Teams
Sources
MoCRA and FDA Regulatory Sources
- National Law Review / Foley and Lardner (May 2026) – How MoCRA Is Reshaping FDA Oversight of Cosmetics in 2026: mandatory registration enforced July 2024; GMP rule pending; talc/asbestos rule withdrawn November 2025; enforcement increasing 2026 and beyond
- Lumanity (May 2026) – FDA Cosmetics Oversight Update for MoCRA: 3x increase in adverse event reports; December 2025 PFAS report (25 PFAS, 0.41% of listed products); Office of Cosmetics moved to Office of Chief Scientist 2024; mandatory recall authority
- Diaz Trade Law (May 2026) – FDA Update Increased Cosmetics Oversight: 15,000 unique active facility registrations; 1M+ product listings; adverse events tripled; Cosmetics Direct portal; enforcement July 1, 2024
- Respect Manufacturing (February 2026) – MoCRA Guide 2025: 15-day adverse event reporting; 6-year record retention (Section 605); safety substantiation and traceability requirements; FDA-registered CGMP facilities
- Bustos Law Group (December 2025) – MoCRA Is Taking Shape: updated FDA timelines; enforcement expectations rising; deadline extensions and core requirements still coming
CAPA, Investigation, and Root Cause Analysis Sources
- ECA Academy (May 2026) – CAPA and Root Cause Analysis: April 2026 warning letters (Active Cosmetics Manufacturing, CareFusion 213); scope extension failure; “after FDA identified gaps” citation; “not a better form but a better system” finding
- ECA Academy (September 2025) – Four Warning Letters on CAPA and Root Cause: Staphylococcus Aureus case study; batch released despite contamination evidence; training and SOP revision deemed insufficient; systematic approach deficiency
- Assyro AI (March 2026) – FDA CAPA Requirements: 64% of FDA enforcement actions 2024 included CAPA deficiencies; top 10 citation every year since 2015; five common deficiency categories; consequences of inadequate CAPA
- Invensis Learning / FDA Group (May 2026) – CAPA Process Explained: CAPA is #1 FDA inspection citation since fiscal year 2010; five common deficiency categories; business benefits of effective CAPA programs
- The FDA Group (January 2025) – Quick Tips for CAPA System Success 2025: compliance mindset vs. problem-solving mindset; risk-based triage; operator and quality collaboration; OOS investigation example; CAPA as trending problem area
- JJC Group (December 2025) – CAPA and Root Cause Analysis Complete Guide: RCA must feed CAPA; inadequate training and resources as CAPA failure cause; continuous improvement through strong RCA culture
- SmartCAPA – Root Cause Analysis: 5 Whys and Fishbone diagram methodology; FMEA and RPN; systematic investigation steps; problem statement documentation
- Center for Professional Innovation and Education (July 2025) – CAPA and Root Cause Analysis: roles in GMP environments; pharmaceutical, biologics, and device CAPA framework; investigation team composition